
Are You Missing These Optic Nerve Disorders?
Understanding the pathophysiology of the anomaly is key to proper diagnosis.
By Justin Cole, OD, and Jarett Mazzarella, OD
Release Date:
February 2017
Expiration Date:
February 15, 2020
Goal Statement:
When clinicians see an abnormal optic nerve, they must wade through a whole host of differentials before reaching the correct diagnosis. This article provides an overview of many common optic neuropathies and the diagnostic testing necessary to hone in on the proper condition so clinicians are ready to tackle the challenge of an unusual optic nerve.
Faculty/Editorial Board:
Justin Cole, OD, and Jarett Mazzarella, OD
Credit Statement:
This course is COPE approved for 2 hours of CE. Course ID is 52257-NO. Check with your local state licensing board to see if this counts toward your CE requirement for relicensure.
Disclosure Statement:
The authors have no relationships to disclose.
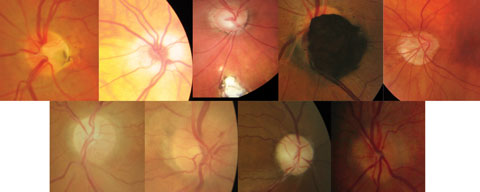 |
| Fig. 1. Collage of non-glaucomatous optic neuropathies. Top (left to right): ONH pit, hypoplastic nerve, ONH/retinal coloboma, melanocytoma, malinsertion. Bottom (left to right): Sectoral pallor, papillitis, optic atrophy, ONH drusen. Click image to enlarge. |
Patient History
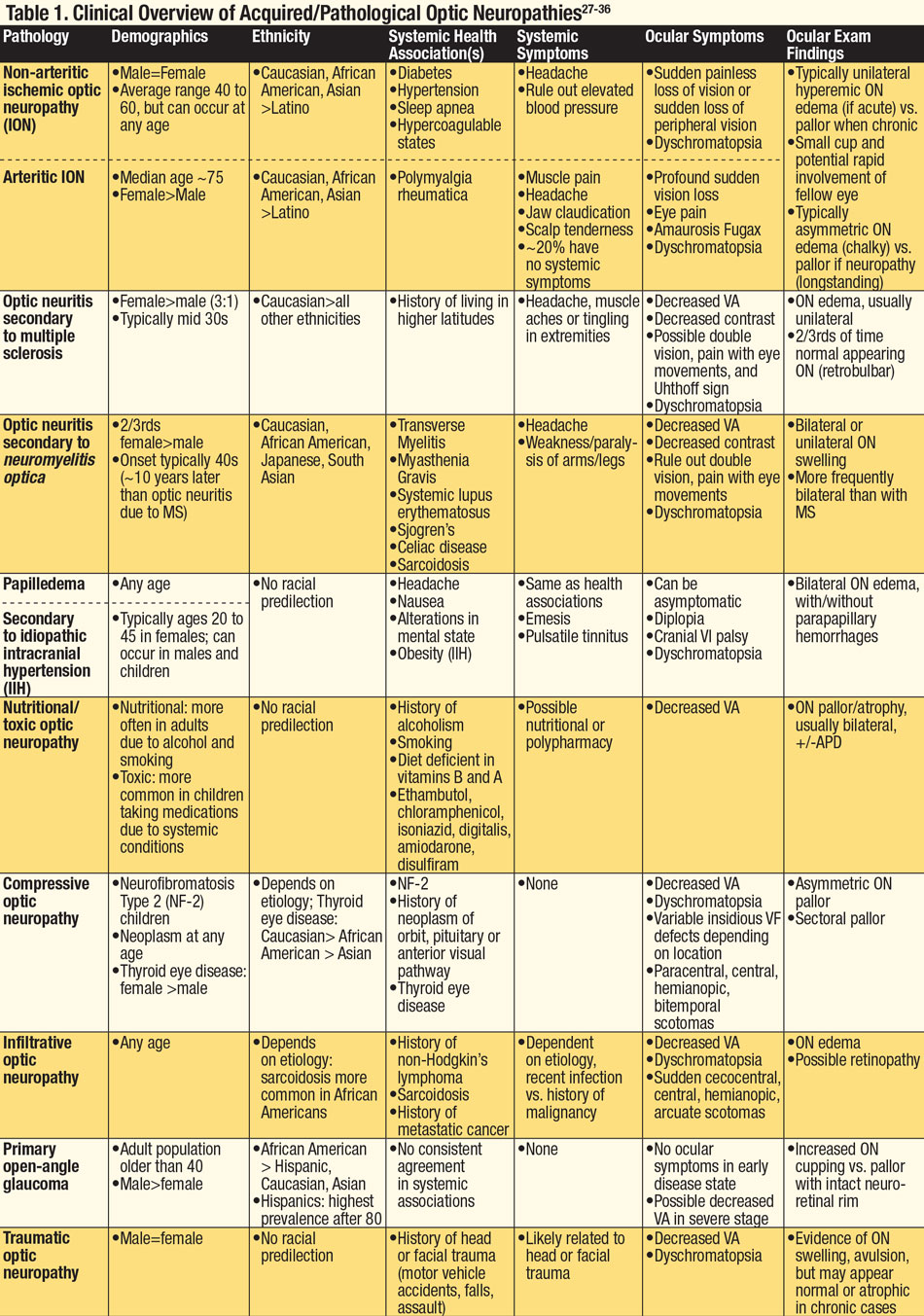
Because many optic neuropathies present with similar findings, the patient’s systemic health, demographics and ethnicity are key to the proper diagnosis. History of associated headaches, changes in mental state, reduced or altered hearing, muscle fatigue, temple pain, history of cancer or malignancy or presence of active infection can all be symptoms of a coexisting systemic condition(s). Knowing which conditions are associated with specific neuropathies can aid the clinician when faced with an abnormal ON. Finally, recognizing whether the anomaly is acquired or congenital and understanding the pathophysiology of the anomaly are integral to determining an accurate diagnosis (Tables 1 and 2).
 |
| Click table to enlarge. |
Diagnostics
Clinicians can rely on several diagnostic strategies to help differentiate acquired vs. congenital optic neuropathies:
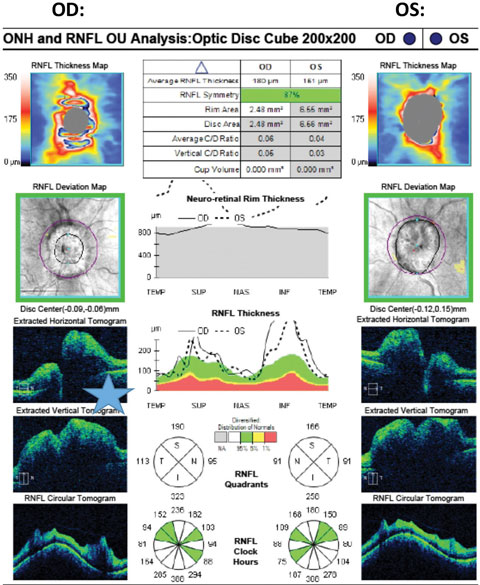 |
| Fig. 2. Cirrus SD-OCT pRNFL scan illustrating significant ONH elevation on B-scans and pRNFL thickening in all quadrants OU. Just superior to the blue star is the retinal nerve interface illustrated in the B-scan, OD, with subretinal fluid causing separation of the RPE layer from the neurosensory retina. |
Pupil testing. This is crucial in determining functional vs. organic etiologies of VA loss, as well as distinguishing the exact cause of the optic neuropathy.2 Research shows a positive association between VF asymmetry from automated perimetry and RAPD measurements.3 However, this is not inclusive of all optic neuropathies, as some diseases of the ON are associated with relative sparing of either pupil function or visual function, as in Leber's optic neuropathy.2
Historically, the swinging flashlight test has been an accurate method of detecting a pupil abnormality, and clinical examination can typically elicit an RAPD sensitivity of three decibels.2 Further accuracy can be obtained with the use of neutral density filters in front of the eye without the RAPD until neutralization of the pupillary response between the eyes is obtained.2 When checking for an RAPD, clinicians must use a very bright light in a dark room to assess the full amplitude of the pupillary response.1 In certain circumstances, anisocoria, reduced contrast due to dark iris color, iris anomalies or injury can lead to erroneous results.4 The lack of light intensity standardization in successive tests can limit the clinician’s ability to identify progression, stability or resolution of pupil findings over time. Also, in the presence of bilateral and symmetric optic neuropathy, an RAPD may be absent.
Recent studies demonstrate that pupillography may be as sensitive in detecting ON disease as other clinical findings such as VA, ganglion cell layer thickness or peripapillary RNFL thickness.5 Pupillography provides more consistent duration and intensity of light compared with manual methods of pupil testing, thereby allowing clinicians to measure a stable and consistent light reflex.4 Studies demonstrate RAPD amplitude scores can detect pupil defects with a high degree of accuracy at any stage of ON disease.6
Color vision testing. Congenital color vision defects are often associated with a cone deficiency.7 In contrast, acquired color vision deficiency occurs after birth and may fluctuate in severity depending on the etiology. When evaluating these patients with color plates, clinicians must assess whether the patient is correct in plate recognition as well as the speed and ease of recognition between eyes in cases where acquired color deficiency is suspected.1 Defects can be difficult to define and may be different from right to left eyes. Acquired color deficiencies can occur from neuropathological events such as stroke or traumatic brain injury, neurodegenerative processes such as Alzheimer’s and multiple sclerosis (MS), certain medications (ethambutol, amiodarone, methotrexate and cyclosporine), or chemicals (carbon monoxide and carbon disulfide).1,7,8
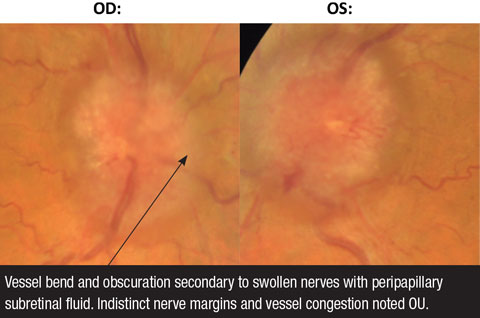 |
| Fig. 3. These fundus photos demonstrate papilledema OD and OS. |
Current research suggests that mechanisms of acquired color vision deficiency due to ON or retinal diseases may be ill-defined. The patterns of acquired dyschromatopsias appear to be heterogeneously associated with the distribution of color vision in the central VF and the particular disease state. For example, patients with ION may have normal color vision if their central field remains intact. Interocular asymmetry and interocular differences in disease manifestations can lead to variances in the color vision defects within and between the two eyes in acquired dyschromotopsia.10
This technology is integral in the primary eye care setting for its ability to detect structural damage in the ON and macula. The clinician is able to detect and quantify the retinal and peripapillary B-scans with a resolution of three to five microns.11 SD-OCT can be applied to identify an optic neuropathy but may be non-specific, not always providing the clinician the exact etiology of the disease state. This technology is also valuable in monitoring disease progression over time and in cases where intervention is prudent, the ability to evaluate the response to treatment.
For example, studies demonstrate that OCT RFNL analysis and ganglion cell-inner plexiform (GC) analysis can detect patients with glaucoma compared with the machine’s normative database (which varies between OCT manufacturers) with a high degree of sensitivity and specificity.12,13 Research on non-glaucomatous neuropathies, such as papilledema, has found a proportion of these patients develop subretinal fluid at the edge of the ON (lazy V sign), which can easily be defined by an OCT line scan (Figures 2 and 3).14 In contrast, Figures 4a and 4b illustrate the lumpy internal contour of the ON and the sharp demarcation at the ON’s edge consistent with ON drusen. These tools can also be used to differentiate papilledema from more benign findings such as vitreopapillary traction, small crowded nerves, ON malinsertion and ON head drusen (Figures 5 and 6).15
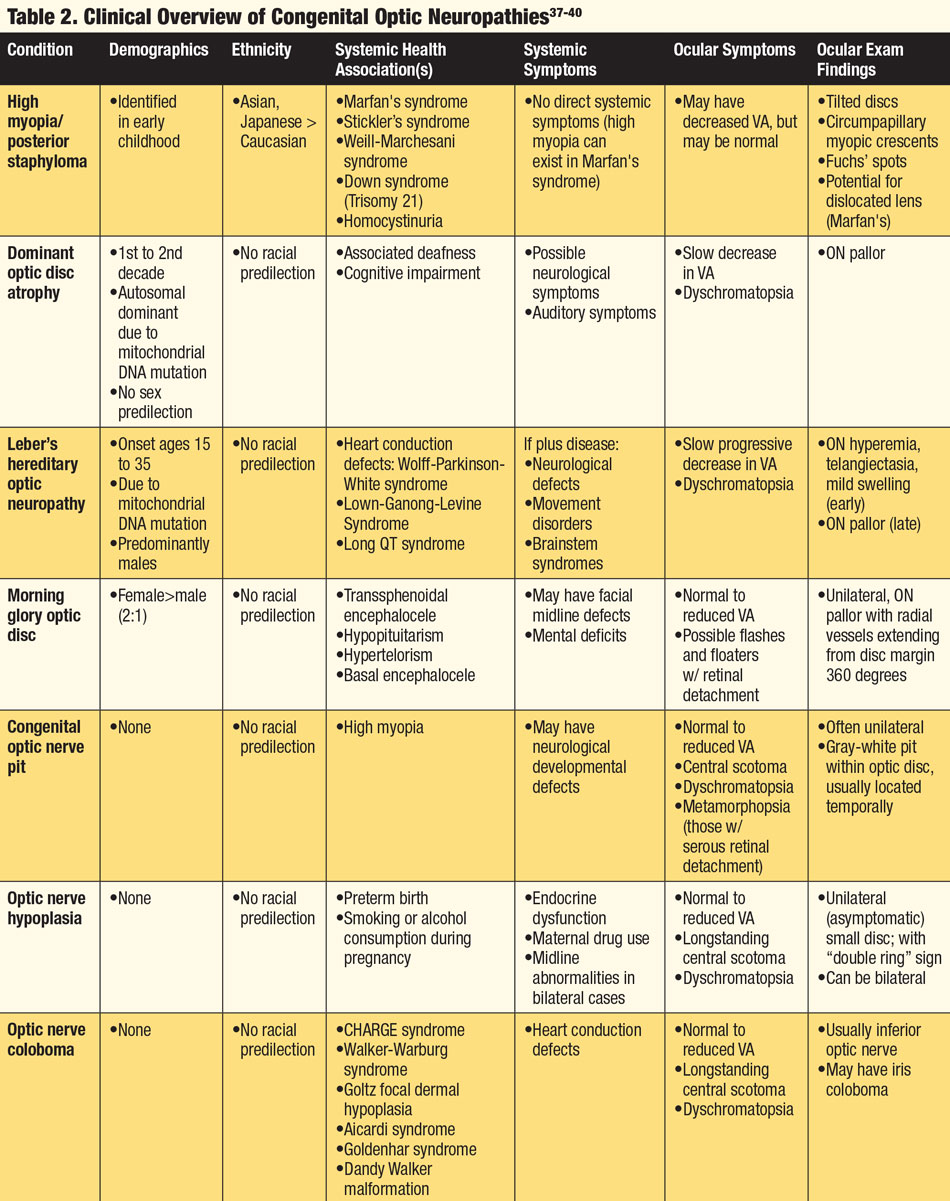 |
| Click table to enlarge. |
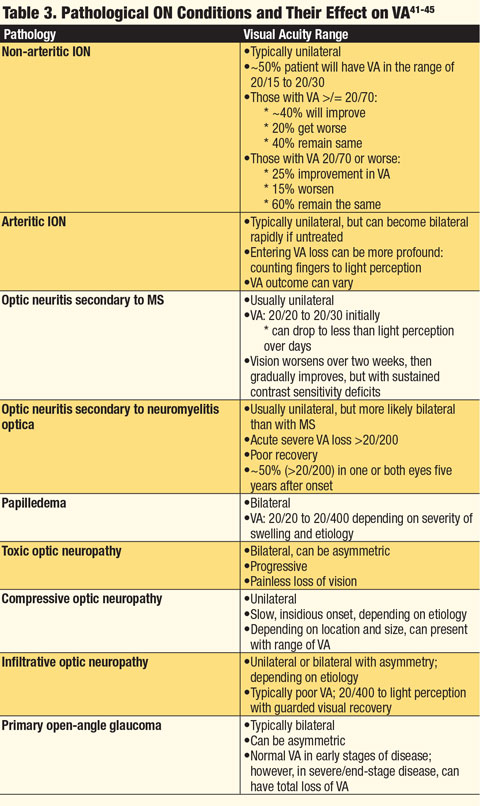 |
| Click table to enlarge. |
While this technology allows early visualization of structural changes, there are limitations. A scan’s reliability can be affected by artifacts, such as media opacities and excessive movement, aberrations due to pupil size, or even the failure of the software to properly define structural boundaries.17 Also, patients are not limited to one disease state, so individuals with concurrent ocular pathologies may have less accurate OCT measurements, specifically in GC analysis.18
Visual field testing. The time of onset of the VF defect and its location can aid in determining the etiology of an optic neuropathy (Table 5). If the onset of the VF loss is sudden, a clinician is more apt to think of inflammatory, ischemic, traumatic or demyelinating etiologies. If the VF loss occurs more gradually over time, the differential should include a compressive lesion, nutritional or toxic neuropathy, inherited neuropathy and glaucoma.1Although VF defects are not specific for a particular optic neuropathy, patterns emerge that can aid in the proper diagnosis. When the location of the defect is cecocentral, toxic, nutritional and hereditary etiologies must be considered. If a clear altitudinal defect exists, ischemic causes should be investigated. If an arcuate or Bjerrum scotoma is found, a glaucomatous etiology should be explored. A defect respecting the vertical meridian should indicate possible lesions at or behind the optic chiasm, and a central defect in one eye with a corresponding “pie in the sky” defect of the opposite eye should elicit prompt workup for a junctional lesion at the chiasm.1
Fundus photography/clinical exam. Retinal fundus photography is useful for documenting normal, swollen, pale or anomalous optic discs, as well as to monitor the ON for structural changes over time.1 Optic disc cupping is often associated with glaucoma, but less commonly may be associated with non-glaucomatous neuropathies.19 Studies have found evaluating the ON rim color, inspecting for rim ablation and correlating these structural clues with the degree of VF loss can differentiate if the optic neuropathy is glaucomatous or non-glaucomatous.20 In glaucoma, cupping is greater than pallor, and pallor typically only occurs in very advanced stages of the disease. When functional field loss is much greater than the structural cupping, the clinician must look at other neurological etiologies.
Finally, in early glaucomatous neuropathy there is a loss of neural rim tissue. In case presentations where pallor is present without rim notching or loss, clinicians should suspect non-glaucomatous neuropathies. In manifestations of optic neuropathy where disc hemorrhages or exudative retinopathy are present, the clinician should consider other etiologies such as ischemic, inflammatory, infectious or infiltrative disease. If the ON looks pale with a chalky appearance, arteritic ION must be considered. In clinical presentations where the ON demonstrates temporal pallor, toxic or nutritional neuropathy needs to be ruled out.1
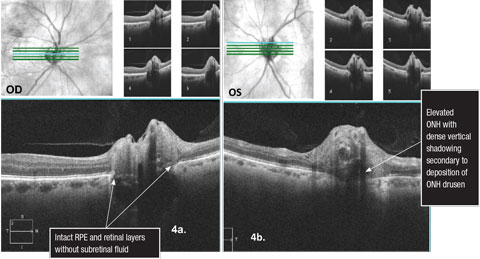 |
| Figs. 4a and 4b. 5 line raster Cirrus SD-OCT scans over the optic nerves illustrating optic nerve head drusen. Click image to enlarge. |
In evaluating the ON for glaucoma, clinicians should follow the ISNT rule—normal ONs typically show a larger rim width Inferior, Superior, Nasal and Temporal. Although this rule is sensitive for glaucoma, it is not specific for a glaucomatous neuropathy.21 Another finding to consider when evaluating the ON for a glaucomatous process is assessing for focal enlargement of the cup, typically greater in the vertical dimension. Excavation of the cup of the ON with the presence of laminar dots, RNFL defects, Drance hemorrhages or beta peripapillary atrophy also may point to a glaucomatous process.22 In non-glaucomatous neuropathy, the ON is typically pale, may have more diffuse rim loss, the field defect does not necessarily correlate with the structural appearance of the ON and peripapillary atrophy is less common.23
In younger patients with normal eye pressure, reduced VA or VF defects respecting the vertical meridian, clinicians should investigate for neurological associations.23
When evaluating any condition that relates to laterality, clinicians should look for comparisons and symmetry. Looking at the non-involved eye to distinguish an anomalous from normal anatomical variation is important. When evaluating the ON, this principle also applies, as tilted discs, ON hypoplasia, past ischemic events or ocular trauma can all lead to an asymmetry between the right and left ON, which can help to further define the etiology of the ocular pathology. Optic nerve size and RNFL thickness should be fairly symmetrical between the two eyes. In cases of larger or smaller appearing ONs, a measurement of the overall disc diameter is prudent.
 |
| Click table to enlarge. |
Lab testing. With unilateral optic disc swelling, a clinician must know when to consider blood work. In an adult population, ION is one of the more common optic neuropathies, and arteritic ION can be indicative of giant cell arteritis (GCA), which can be fatal. Therefore, clinicians should obtain a complete blood count (CBC), c-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) in cases of suspected arteritic ION based on clinical assessment. Knowing the expected normative values of these labs is also important. For ESR, the expected range is equal to the age of a male patient divided by two and the age of a female patient +10 divided by two. Elevated ESR, CRP and thrombocytosis (elevated platelet count) are positive predictors of GCA. Studies show elevated levels of CRP and the presence of thrombocytosis may be indicative of arteritic ION, even when the ESR is not elevated. When all three lab values are elevated, the likelihood of a positive temporal artery biopsy due to occult GCA is eight times more likely.24
Unilateral morning glory syndrome, ON hypoplasia, ON pits, ON coloboma, myopic/tilted discs and ON drusen generally don’t require laboratory testing, as they are a clinical diagnosis.
In cases of unilateral ON pallor, and especially those with contralateral optic nerve swelling, and the presence of VF loss, neuroimaging of the head and orbits is indicated to rule out compressive or intracranial etiologies.
 |
| Click table to enlarge. |
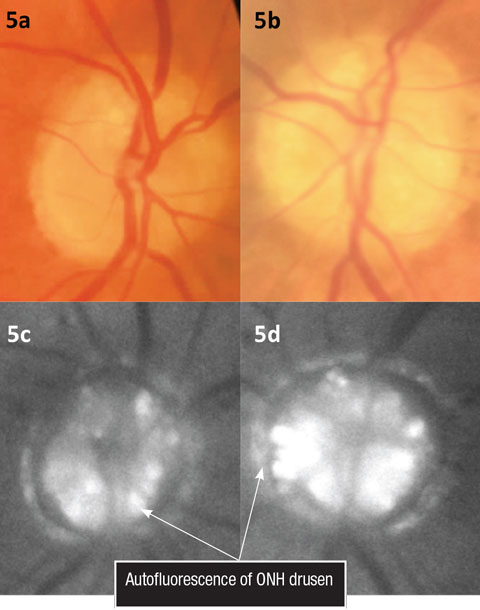 |
| Figs. 5a and 5b. Color fundus photos illustrating ONH drusen. Figs. 5c and 5d. Fundus autofluorescence of the optic nerves. Click image to enlarge. |
Bilateral ON edema, symmetrical or asymmetrical, is an ocular emergency. Differential diagnoses must be explored based on history, and neuroimaging, specifically an MRI of the head with contrast, is warranted. In suspected cases, magnetic resonance venography may also be completed to rule out sinus thrombosis. In cases where intracranial etiologies are ruled out by negative neuroimaging, a lumbar puncture can confirm the diagnosis of idiopathic intracranial hypertension when suspected.25In other optic neuropathies, the clinical presentation can vary; in toxic, infiltrative or infectious etiologies it is usually bilateral; however, it can assume an asymmetric appearance. Patients with suspected malignant hypertensive retinopathy or diabetic papillitis should have their blood pressure evaluated and their hemoglobin A1c checked. Clinicians should order a CBC with differential in cases with a history or suspicion of blood dyscrasias or leukemia. During the case history, any radiation to the face, orbit or adnexa within the last five years should be addressed, as there can be delayed ocular sequelae. Referral to the primary care provider is necessary in suspected cases of ocular lymphoma or metastatic disease.
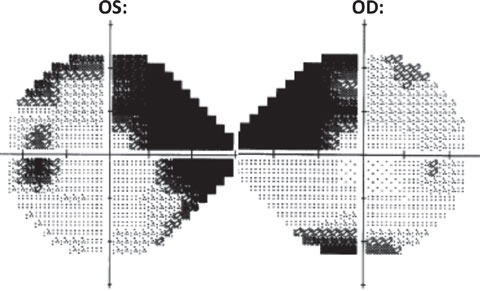 |
| Fig. 6. HVF 24-2 grayscale: OD (right) shows a dense superior nasal step with inferior defect. OS (left) shows a dense superior nasal step with associated incomplete arcuate formation as well as a dense inferior nasal step. Also noted OS is an enlarged blind spot. |
Bilateral optic neuropathies tend to have the worst visual consequence. Leber’s hereditary optic neuropathy and dominant optic atrophy tend to occur early in life, and in some cases a cardiac workup is advised secondary to associated heart block. Lab testing for the presence or absence of the main mitochondrial mutations in Leber’s accounting for most cases can be performed to confirm the diagnosis.26 In all cases of hereditary optic neuropathy, the visual prognosis is guarded, and genetic counseling may be considered for the patient and their family.27
Optic neuropathies may manifest in various presentations, and as primary eye care providers, it is crucial to identify ocular conditions that can have longstanding health and visual consequences. By possessing the knowledge and the tools to identify the ON abnormality, clinicians can arrive at the correct diagnosis and provide timely and appropriate management.
Drs. Cole and Mazzarella are staff optometrists in the VA Health Care System, Salisbury, NC.
|
1. Behbehani R. Clinical approach to optic neuropathies. Clinical Ophthalmology (Auckland, NZ). 2007;1(3):233-46. 2. Bremner FD. Pupil assessment in optic nerve disorders. Eye (Lond.). 2004;18(11):1175-81. 3. Brown RH, Zilis JD, Lynch MG, Sanborn GE. The afferent pupillary defect in asymmetric glaucoma. Arch Ophthalmol. 1987;105(11):1540-3. 4. Konan Medical. RAPDx high-definition pupil diagnostics. Available at http://konanmedical.com/rapdx. Accessed October 31, 2016. 5. Takizawa G, Miki A, Maeda F, et al. Association between a relative afferent pupillary defect using pupillography and inner retinal atrophy in optic nerve disease. Clin Ophthalmol. 2015;9:1895-903. 6. Thompson HS, Montague P, Cox TA, Corbett JJ. The relationship between visual acuity, pupillary defect, and visual field loss. Am J Ophthalmol. 1982;93(6):681-8. 7. Pacheco-Cutillas M, Edgar DF, Sahraie A. Acquired colour vision defects in glaucoma-their detection and clinical significance. Br J Ophthalmol. 1999;83(12):1396-402. 8. Colour Blind Awareness. Aquired colour vision defects. Available at www.colourblindawareness.org/colour-blindness/acquired-colour-vision-defects. Accessed October 31, 2016. 9. Simunovic MP. Acquired color vision deficiency. Surv Ophthalmol. 2016;61(2):132-55. 10. Hart WM Jr. Acquired dyschromatopsias. Surv Ophthalmol. 1987;32(1):10-31. 11. Kiernan DF, Mieler WF, Hariprasad SM. Spectral-domain optical coherence tomography: a comparison of modern high-resolution retinal imaging systems. Am J Ophthalmol. 2010;149(1):18-31. 12. Mansouri K, Leite MT, Medeiros FA, et al. Assessment of rates of structural change in glaucoma using imaging technologies. Eye (Lond.). 2011;25(3):269-77. 13. Sung KR, Wollstein G, Kim NR, et al. Macular assessment using optical coherence tomography for glaucoma diagnosis. Br J Ophthalmol. 2012;96(12):1452-5. 14. Tawse KL, Hedges TR 3rd, Gobuty M, Mendoza-Santiesteban C. Optical coherence tomography shows retinal abnormalities associated with optic nerve disease. Br J Ophthalmol. 2014;98 Suppl 2:ii30-3. 15. Lee TH, Heo H, Park SW. Clinical usefulness of spectral-domain optical coherence tomography in glaucoma and NAION. Chonnam Med J. 2016;52(3):194-200. 16. London A, Benhar I, Schwartz M. The retina as a window to the brain-from eye research to CNS disorders. Nature Reviews Neurology. 2013;9(1):44-53. 17. Somfai GM, Salinas HM, Puliafito CA, Fernandez DC. Evaluation of potential image acquisition pitfalls during optical coherence tomography and their influence on retinal image segmentation. J Biomedical Optics. 2007;12(4):041209. 18. Arintawati P, Sone T, Akita T, et al. The applicability of ganglion cell complex parameters determined from SD-OCT images to detect glaucomatous eyes. Journal of glaucoma. 2013;22(9):713-8. 19. Ambati BK, Rizzo JF 3rd. Nonglaucomatous cupping of the optic disc. Internat Ophthalmol Clinics. 2001;41(1):139-49. 20. Zhang YX, Huang HB, Wei SH. Clinical characteristics of nonglaucomatous optic disc cupping. Exper Thera Med. 2014;7(4):995-9. 21. Pogrebniak AE, Wehrung B, Pogrebniak KL, et al. Violation of the ISNT rule in nonglaucomatous pediatric optic disc cupping. Invest Ophthalmol Vis Sci. 2010;51(2):890-5. 22. Jonas JB, Budde WM. Diagnosis and pathogenesis of glaucomatous optic neuropathy: morphological aspects. Progress in Retinal and Eye Research. 2000;19(1):1-40. 23. Lee AG. Differentiating glaucomatous from nonglaucomatous optic atrophy. Ophthalmology. 1999;106(5):855. 24. Walvick MD, Walvick MP. Giant cell arteritis: laboratory predictors of a positive temporal artery biopsy. Ophthalmology. 2011;118(6):1201-4. 25. Friedman DI. Papilledema and idiopathic intracranial hypertension. Continuum. 2014;20(4 Neuro-ophthalmology):857-76. 26. Yu-Wai-Man P, Chinnery PF. Leber hereditary optic neuropathy. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews. Seattle: University of Washington, Seattle; 1993. 27. Wu CY, Niziol LM, Musch DC, Kahana A. Thyroid-related orbital decompression surgery: a multivariate analysis of risk factors and outcomes. Ophthal Plast Reconstr Surg. 2016 Apr 19. [Epub ahead of print]. 28. Cestari DM, Gaier ED, Bouzika P, et al. Demographic, systemic, and ocular factors associated with nonarteritic anterior ischemic optic neuropathy. Ophthalmology. 2016;123(12):2446-55. 29. Skorin L Jr., Knutson R. Ophthalmic diseases in patients with obstructive sleep apnea. J Am Osteopathic Assoc. 2016;116(8):522-9. 30. Chen JJ, Leavitt JA, Fang C, et al. Evaluating the incidence of arteritic ischemic optic neuropathy and other causes of vision loss from giant cell arteritis. Ophthalmology. 2016;123(9):1999-2003. 31. Pau D, Al Zubidi N, Yalamanchili S, et al. Optic neuritis. Eye (Lond.). 2011;25(7):833-42. 32. Trebst C, Jarius S, Berthele A, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurology. 2014;261(1):1-16. 33. Wingerchuk DM, Lennon VA, Lucchinetti CF, et al. The spectrum of neuromyelitis optica. The Lancet Neurology. 2007;6(9):805-15. 34. Kaiser PK, Friedman N. The Massachusetts Eye and Ear Infirmary Illustrated Manual of Ophthalmology. 3rd Ed. Philadelphia: Elsevier; 1998. 35. Orssaud C, Roche O, Dufier JL. Nutritional optic neuropathies. J Neurological Sciences. 2007;262(1-2):158-64. 36. Hayreh SS, Podhajsky PA, Zimmerman B. Occult giant cell arteritis: ocular manifestations. Am J Ophthalmol. 1998;125(4):521-6. 37. Fraser JA, Biousse V, Newman NJ. The neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol. 2010;55(4):299-334. 38. Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. 2005;140(3):517-23. 39. Dutton GN. Congenital disorders of the optic nerve: excavations and hypoplasia. Eye (Lond.). 2004;18(11):1038-48. 40. Logan NS, Gilmartin B, Marr JE, et al. Community-based study of the association of high myopia in children with ocular and systemic disease. Optom Vis Sci. 2004;81(1):11-3. 41. Hayreh SS. Ocular vascular occlusive disorders: natural history of visual outcome. Progress in Retinal and Eye Research. 2014;41:1-25. 42. Costello F. Inflammatory optic neuropathies. Continuum. 2014;20(4 Neuro-ophthalmology):816-37. 43. Wingerchuk DM, Weinshenker BG. Neuromyelitis optica: clinical predictors of a relapsing course and survival. Neurology. 2003;60(5):848-53. 44. Chiotoroiu SM, Noaghi M, Stefaniu GI, et al. Tobacco-alcohol optic neuropathy--clinical challenges in diagnosis. J Med Life. 2014;7(4):472-6. 45. Wong D, Danesh-Meyer H, Pon JA. Infiltrative lymphomatous optic neuropathy in non-Hodgkin lymphoma. J Clin Neuroscience. 2015;22(9):1513-5. 46. Ceynowa DJ, Wickstrom R, Olsson M, et al. Morning glory disc anomaly in childhood - a population-based study. Acta Ophthalmologica. 2015;93(7):626-34. 47. Georgalas I, Ladas I, Georgopoulos G, Petrou P. Optic disc pit: a review. Graefe's Arch Clin Exp Ophthalmol. 2011;249(8):1113-22. 48. Garcia-Filion P, Borchert M. Optic nerve hypoplasia syndrome: a review of the epidemiology and clinical associations. Curr Treat Opt Neurol. 2013;15(1):78-89.1. |







